Introduction#
We’re proud to introduce scvi‑tools v1.4, encompassing major advances in modeling, data loading, computational scalability, metric integration, and interpretability in single-cell analytics.
Featuring nine new or enhanced models, optimized for spatial, cytometry, methylation, perturbation, and multi‑omic data, it also introduces custom data loaders for large-scale datasets, multi‑GPU model training, on-the-fly metric tuning, and integrated model interpretability.
This article delves into each enhancement with depth, including detailed insights, illustrative figures, and manuscript references.
1. 🔬 New Models#
ResolVI#
ResolVI1 is a spatial transcriptomics denoising model that reallocates mis-assigned gene counts among true cells, neighborhood leakage, and background. It employs a Gaussian-mixture latent prior to learn corrected counts and interpretable embeddings. This approach is highly scalable (handling >1 million spots) and offers downstream capabilities like differential expression and transfer learning on corrected data
In tutorial, it has been shown to markedly enhance spatial expression accuracy in noisy segmentation settings, enabling reliable differential expression especially in high-throughput ST datasets.
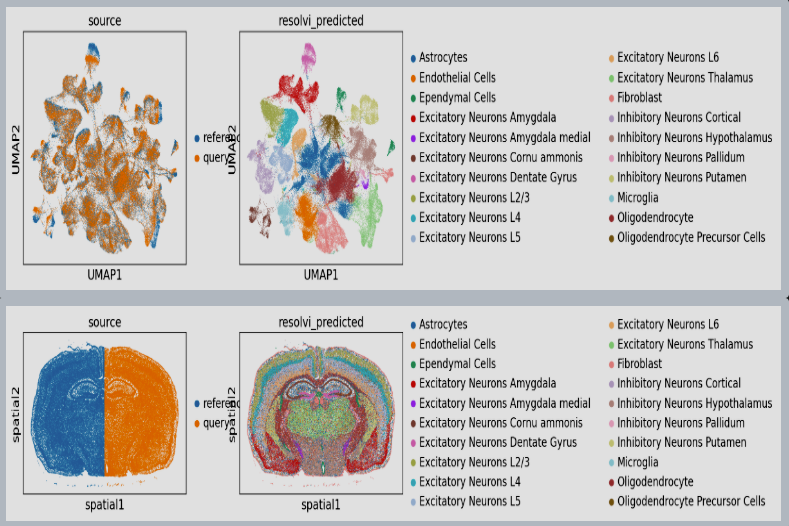
Figure 1: ResolVI cell type annotations based on noisy cellular segmentation Xenium data of a mouse brain. The left hemisphere for model training and the right hemisphere for transfer mapping.
scVIVA#
scVIVA2 augments spatial transcriptomics analysis by jointly modeling each cell’s own expression and its micro-environmental context (neighborhood composition and gene counts). This niche-aware VAE embeds both cellular identity and environmental features, revealing tissue-specific patterns and environment-driven variation. Its latent embeddings delineate tissue-specific structures - ideal for spatial differential abundance or niche-focused clustering studies.
Dedicated tutorial showcase how scVIVA enables niche-focused clustering and differential abundance analyses
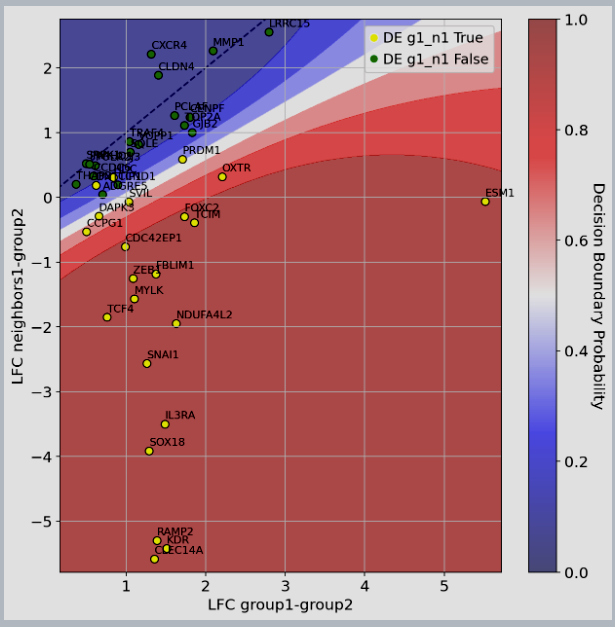
Figure 2: scVIVA results: median Log-Fold Change (LFC) of upregulated genes in vs displayed on the x-axis, while we compare differential expression computed between and on the y-axis. Genes are colored by their marker label (yellow=significantly upregulated in vs , green otherwise). We also display the classifier decision boundary (the predicted probability of being in the yellow class).
CytoVI#
CytoVI3 brings totalVI-inspired modeling to cytometry and mass cytometry data. It models protein-marker distributions, corrects for dropouts and technical batch variation, and generates embeddings for downstream clustering and abundance inference.
Early tutorial already demonstrate clear delineation of immune subpopulations across batch-affected datasets.
VIVS#
Variational Inference for Variable Selection (VIVS4) identifies associations across modalities such as gene–protein couplings—while rigorously controlling false discovery rates using conditional randomization. VIVS achieves interpretable and scalable feature selection, enabling discovery of biologically meaningful links in paired datasets
Tutorial in scvi-tools soon to be updated.
SysVI#
SysVI5 tackles major batch effects, such as those arising from cross-species or organoid-versus-tissue studies—using latent cycle-consistency and VampPrior regularization. Compared to Harmony or regular scVI models, SysVI excels at aligning technical systems while preserving true biological variance, producing embedding spaces where analogous cell types across batches cluster coherently.
in the tutorial, we show the power of sysVI with data integration between human and mouse immune cells.
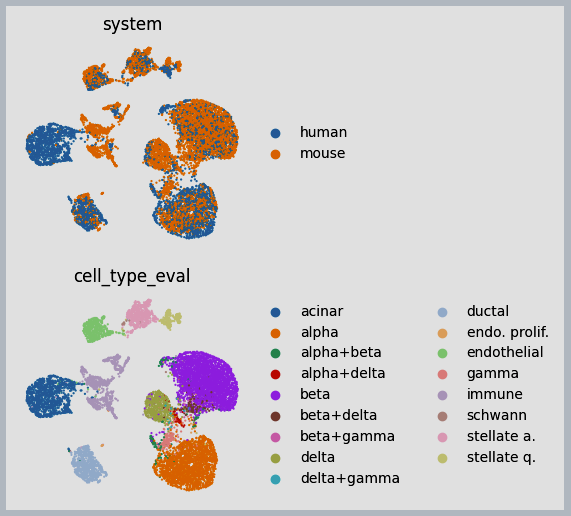
Figure 3: Example results of integration between human and mouse immune cells
Decipher#
Decipher6 Designed to dissect perturbation effects (e.g., disease versus control), Decipher disentangles shared and condition-specific variation within a VAE framework. Demonstrated on AML (acute myeloid leukemia) data, it uncovers latent axes aligning with known disease signatures and identifies corresponding differentially expressed markers - bridging latent space analysis and functional biology.
MethylVI#
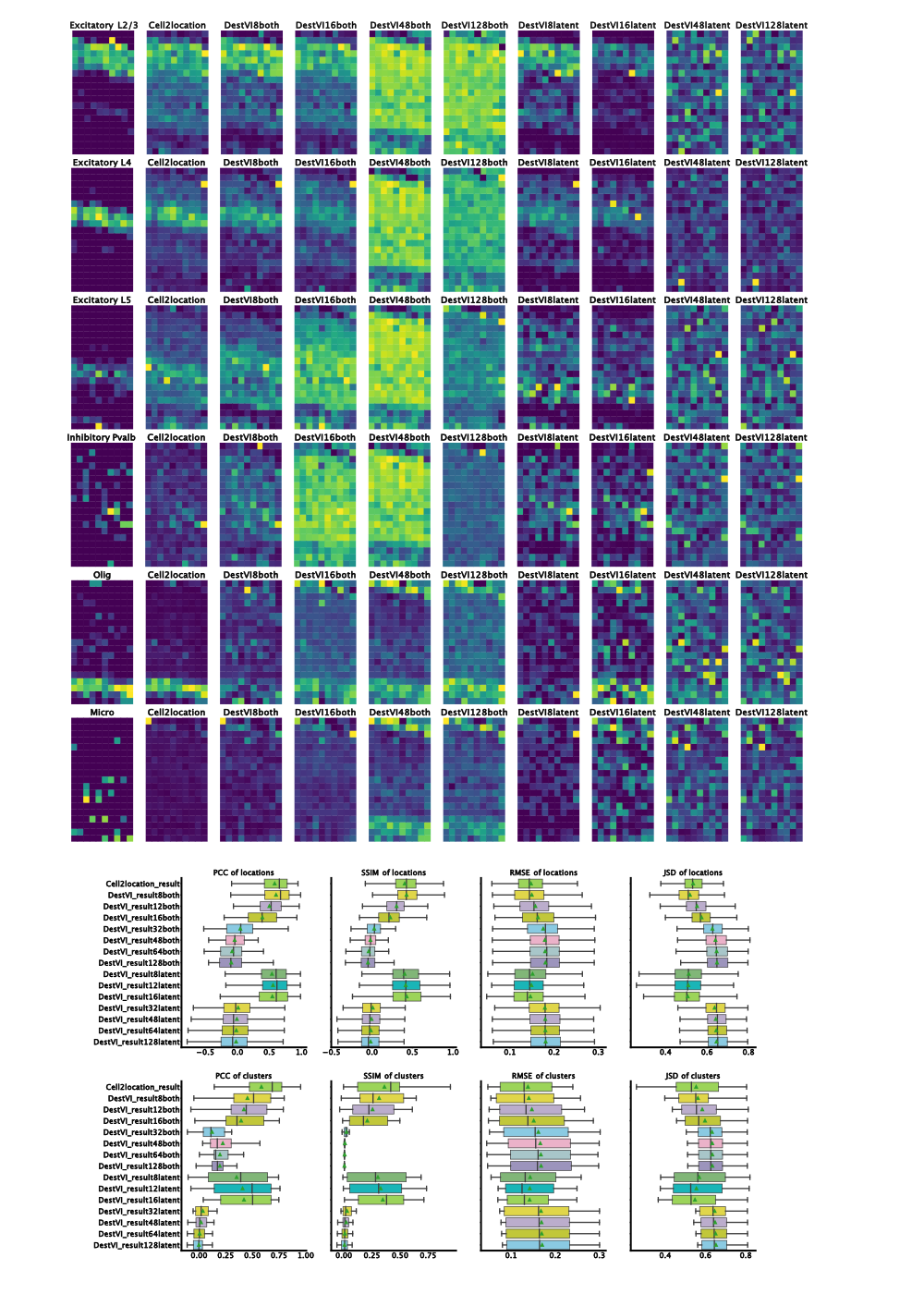
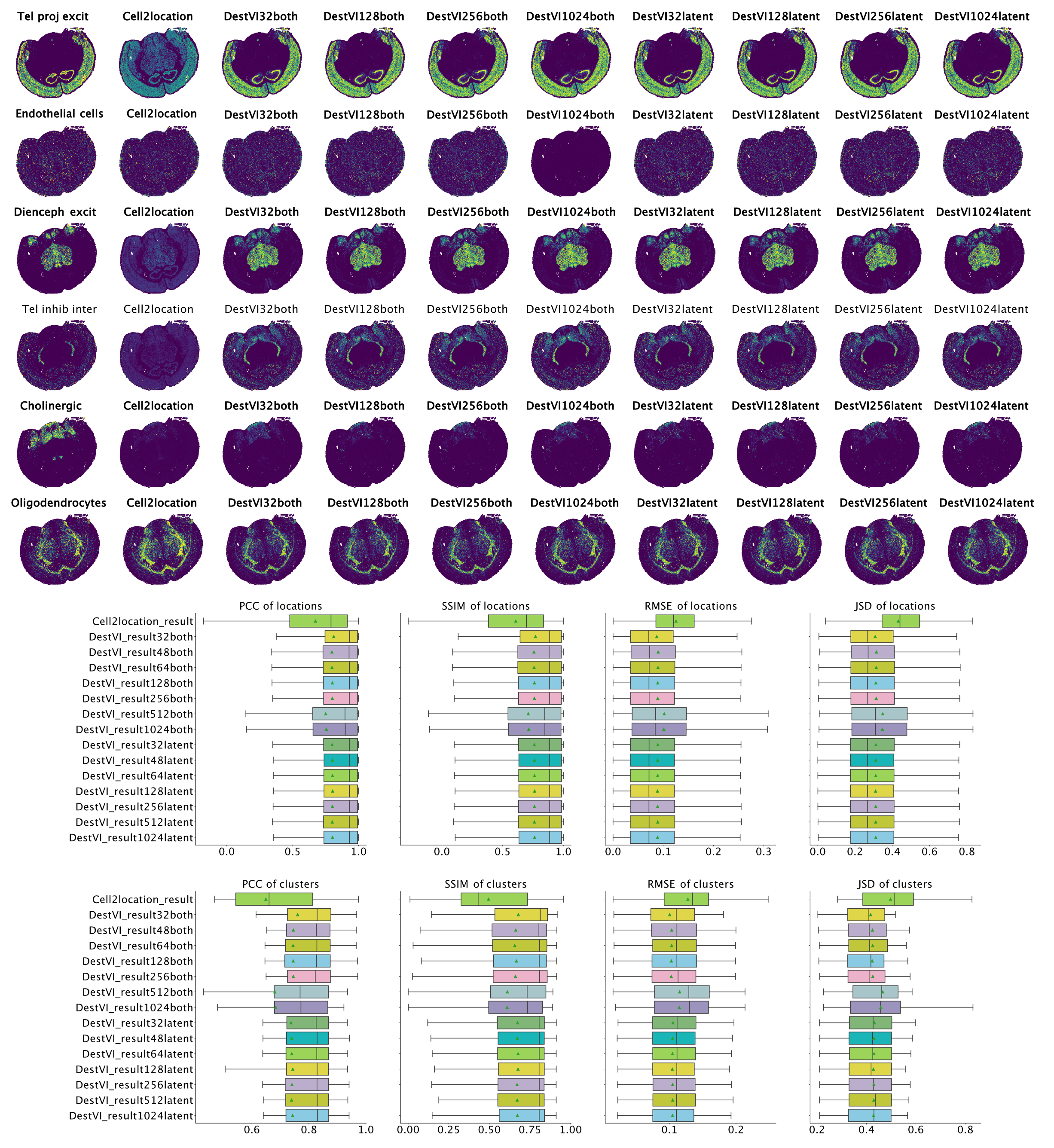
MethylVI7 is a VAE tailored for single-cell bisulfite sequencing (scBS‑seq). By modeling methylation probabilities at genomic regions, it captures epigenetic heterogeneity and learns latent spaces that integrate multiple batches. Tutorial show it outperforms linear methods like PCA in retaining biologically meaningful methylation structures.
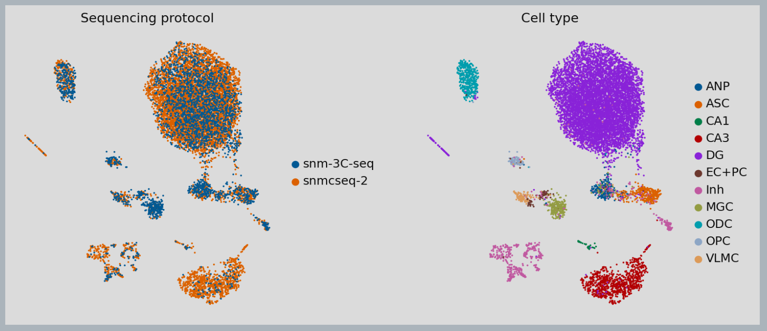
Figure 4: MethylVI integration of cell types from different single-cell bi-sulfite sequencing platforms
MethylANVI#
MethylANVI extends the MethylVI framework with annotation-aware modeling: it jointly integrates methylation profiles with metadata-driven cell-type labels. This supervised model supports both clustering and label transfer, all while capturing latent biological variation across methylome profiles, ideal for epigenetic atlas-building.
totalANVI#
totalANVI brings supervised annotation to CITE‑seq-style multi-omic integration. Leveraging a VAE for RNA + protein, it jointly learns latent embeddings and cell-type classifiers. The model simultaneously performs dimensionality reduction, denoising, differential expression, and accurate cell-type annotation in one cohesive model.
Tutorial in scvi-tools soon to be updated.
mrVI in pyTorch#
MrVI8 (Multi-resolution Variational Inference) is a model written in Jax for analyzing multi-sample, multi-batch single-cell RNA-seq data. MrVI is particularly suited for single-cell RNA sequencing datasets with comparable observations across many samples. It conducts both exploratory analyses (locally dividing samples into groups based on molecular properties) and comparative analyses (comparing pre-defined groups of samples in terms of differential expression and differential abundance) at single-cell resolution.
In recent scvi-tools releases we added a Pytorch implementation of mrVI, to be along with the Jax one, to support broader options on how to use this popular model.
Tutorial of mrVI in torch running on a subset of Tahoe100M cells dataset can be found here
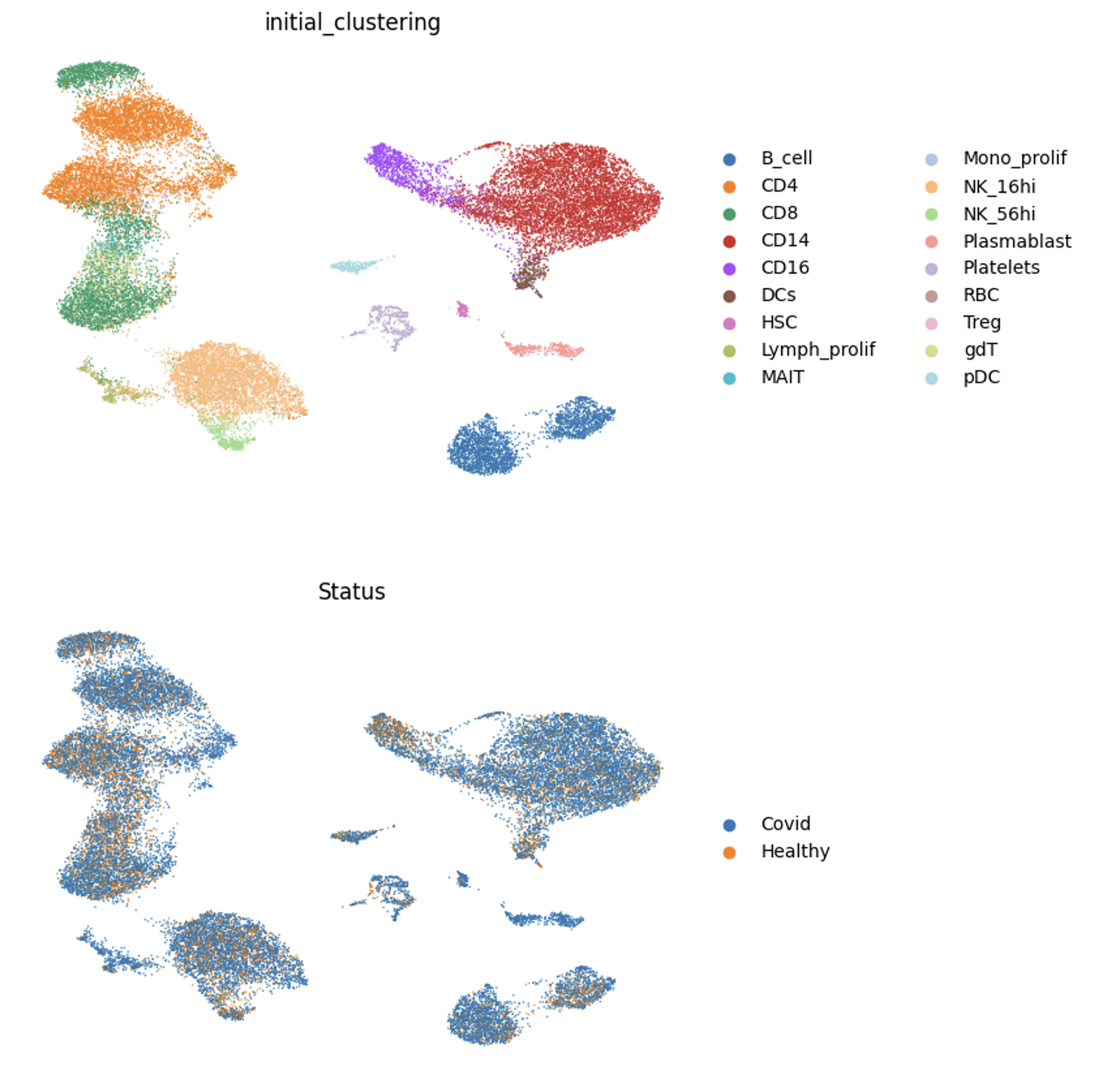
Figure 5: mrVI Integration on Covid dataset
2. 🧩 Custom Dataloaders#
scvi‑tools v1.4 introduces three scalable custom dataloaders: LaminDB, Census, and AnnCollection, enabling out-of-core and federated training without memory overload. Custom Dataloaders are only supported in SCVI & SCANVI models, but it should be easy to expand them to other models. These backends support full compatibility with scvi‑tools data registration and training workflows, offering both scale and convenience to large projects.
LaminDB#
Integrates with Lamindb, enabling out-of-core training from disk-backed collections. Users can register collections and seamlessly train models like SCVI using lamin's MappedCollection, benefiting from disk efficiency while maintaining full API compatibility with in-memory datasets. For more information see this link.
The next tutorial shows demonstration of a scalable approach to training an scVI model on PBMC data using Lamin dataloader.
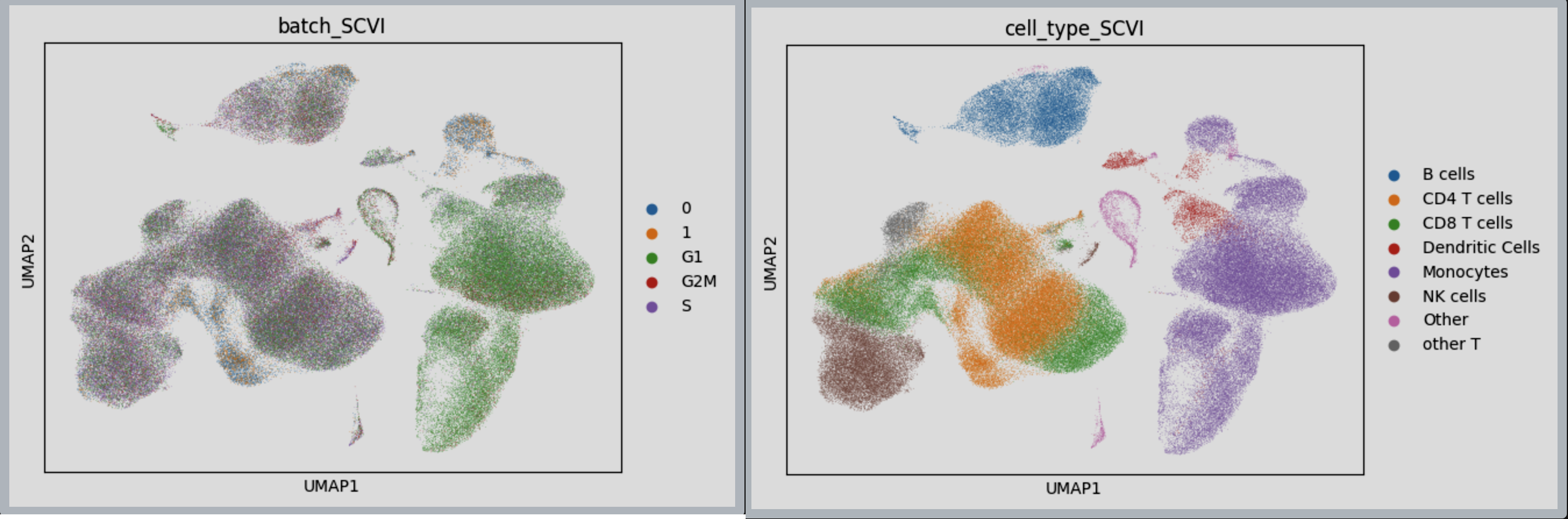
Figure 6: SCVI Integration achieved using LaminDB dataloader, on 2 distinct PBMC data.
This tutorial shows the analysis of mrVI in its PyTorch version together with Lamin Custom dataloader over a subset of Tahoe100M cells dataset.

Figure 7: SCVI (bottom) & MRVI (top) Integration achieved using LaminDB dataloader, on a subset of Tahoe100M cells data.
Census#
employs TileDB-SOMA for atlas-scale tensor-backed data, offering similar streaming capabilities but enhanced support for multidimensional genomic inputs and federated study designs. This custom dataloader directly read cellXgene dataset from S3 and train the SCVI model without the need to first download it, thus very suitable for few shots learning.
The next tutorial shows demonstration of a scalable approach to training an scVI model on mus_musculus data using the Census dataloader

Figure 8: SCVI Cell Integration achieved using Census dataloader, based on 4 type of batches: dataset_id, donor_id, assay and tissue_general
AnnCollection#
This dataloader allows training on multiple AnnData objects simultaneously, without merging them into one dataset. AnnCollection handles disparities in features or layers internally and aligns them during training, empowering federated or multi-study analyses
The next tutorial shows how to apply the annCollection wrapper in scvi-tools to load and train SCANVI model on several adata's that are stored on disk. Another link shows how the Tahoe100M cells dataset was trained in SCVI using the annCollection wrapper and its minified version was stored on scvi-hub for further analysis.
3. ⚙️ Core Enhancements#
Multi‑GPU Training#
Built on PyTorch Lightning, v1.4 empowers all major models to run across multiple GPUs with a single API flag. Training benchmarks times reduced by number of GPU exists, with full gradient synchronization and no code modifications needed. See the following tutorial and info page
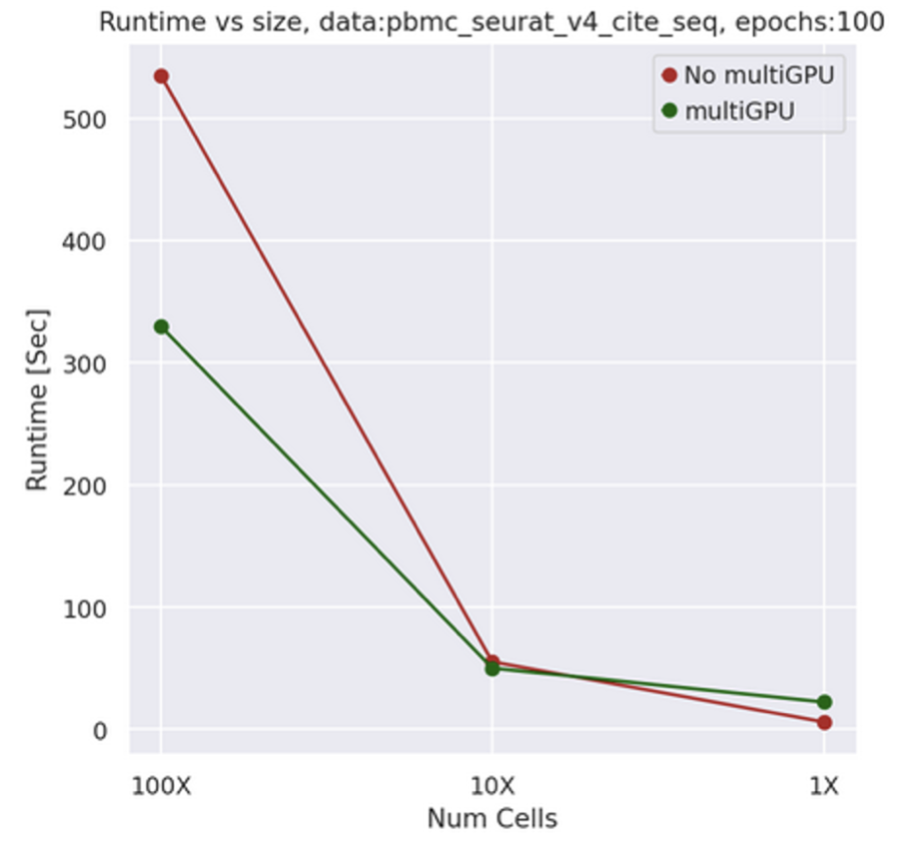
Figure 9: comparison of SCVI training time between single and X2 multi-GPU machines as data increase.
scIB‑Metrics Optimization#
With the integration of ScibCallback and the AutotuneExperiment class, users can now monitor scIB metrics on the validation set during training and automatically tune hyperparameters (model, training and architecture parameters) based on these metrics—directly optimizing for clustering and batch mixing performance, making the model training process more principled and outcome-driven.
See the following tutorial
Explainability & Interpretability#
v1.4 brings native support for Captum's Integrated Gradients (IG) across semi-supervised generative models like Scanvi and totalANVI. Users can compute marker-level attribution scores tied to latent dimensions or differential axes. This complements get_normalized_expression, delivering a full pipeline that links model representations back to biologically interpretable molecular mechanisms, offering transparency and interpretability in deep generative modeling
See the following tutorial as an example of scanvi model ran on a PBMC dataset from 10X.
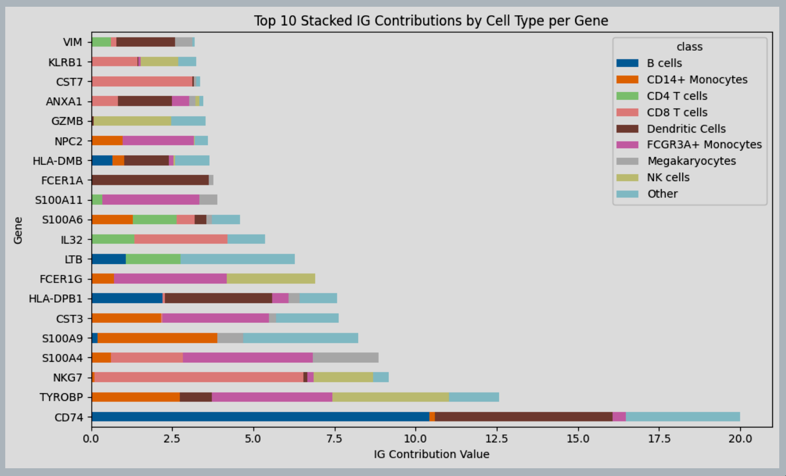
Figure 10: Integrated gradients total contribution per gene per cell type, over data of PBMC.
Summary#
scvi‑tools v1.4 is a landmark release that advances the field across three foundational pillars:
Innovative modeling across ten tailored VAEs for spatial, protein, methylation, perturbation, and multi‑omic data.
Scalable data processing with three new custom dataloaders in the backend, enabling efficient handling of federated, out-of-core, atlas-scale datasets, such as the Tahoe100M cells.
Infrastructure and transparency with multi-GPU training, metric-aware tuning, and demonstrable model interpretability.
Together, these developments empower researchers to build, train, and interpret probabilistic models at scale-in a reproducible, transparent, and biologically meaningful way.
References#
- ResolVI: addressing noise and bias in spatial transcriptomics / Ergen et al.↩
- scVIVA: a probabilistic framework for representation of cells and their environments in spatial transcriptomics / Levy et al.↩
- CytoVI: Deep generative modeling of antibody-based single cell technologies / Ingelfinger et al.↩
- VI-VS: calibrated identification of feature dependencies in single-cell multiomics / Boyeau et al.↩
- sysVI: Integrating single-cell RNA-seq datasets with substantial batch effects / Hrovatin et al.↩
- Decipher: Joint representation and visualization of derailed cell states with Decipher / Nazaret et al.↩
- MethylVI: A deep generative model of single-cell methylomic data / Weinberger et al.↩
- MrVI: Deep generative modeling of sample-level heterogeneity in single-cell genomics / Boyeau et al.↩